





The clinical manifestations of gout are due to interactions between monosodium urate (MSU) crystals and local tissues. This review article outlines recent advances in the understanding of the mechanisms of inflammation in gout. We focus on the cellular response to MSU crystals during acute arthritis, termination of the acute attack and maintenance of asymptomatic hyperuricaemia, and chronic tophaceous disease. We then propose a unifying model of gout involving the differential role of mononuclear phagocytes in the regulation of the inflammatory response to MSU crystals.
The acute gout attack
Initiation of the acute gout attack
The acute attack of gout has all the hallmarks of an acute inflammatory response. Histological examination of the synovium in acute gout shows lining layer hyperplasia and intense infiltration of the membrane by neutrophils, mononuclear phagocytes and lymphocytes [1, 2]. Acute attacks of gout are often triggered by specific events, such as trauma, surgery, intercurrent illness, excess alcohol intake or drugs that alter serum urate levels. Such events may stimulate de novo formation of MSU crystals or may trigger release of microcrystals from preformed deposits within the joint. Although supersaturation of interstitial fluid with MSU is required for the development of crystals, other factors, such as local temperature and pH within the joint, also influence whether crystal formation occurs [3]. Debris within the synovial cavity may provide an initial nucleus for early crystal development [4]. In addition, MSU crystal nucleation may be stabilized by albumin and by immunoglobulin [5, 6]. Depletion of endogenous mast cells has been found to significantly inhibit neutrophil influx in the murine MSU crystal-induced peritonitis model, suggesting a role for crystal-induced mast cell degranulation in initiating inflammation [7]. Mast cells contain preformed proinflammatory substances, including histamine, cytokines [including tumour necrosis factor α (TNF-α)] and enzymes such as tryptase, mast cell chymase and serine esterases, all of which may contribute to the promotion of downstream inflammatory cascades.
Leucocyte recruitment
A central component of acute inflammation involves the activation of vascular endothelial cells, leading to vasodilatation with increased blood flow, increased permeability to plasma proteins and the recruitment of leucocytes into the tissues. Initial endothelial activation with expression of adhesion molecules such as E-selectin, intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) may be caused by factors such as TNF-α released by mast cells [8] (see above), and it is likely that endothelial activation is then amplified by factors released by leucocytes entering the tissues and encountering crystals. Experiments using human umbilical vein endothelial cells have shown that MSU-stimulated monocyte supernatants induce expression of E-selectin, ICAM-1 and VCAM-1, and that this effect is entirely attributable to release of TNF-α and interleukin (IL)-1β [9]. Moreover, in a pig model of MSU-induced arthritis, blockade of TNF-α significantly inhibited E-selectin expression and neutrophil recruitment [9]. Leucocyte recruitment is also likely to be enhanced by the local generation of chemotactic factors such C5a, S100A8/A9 and IL-8 [10, 11]. In a rabbit model of MSU crystal-induced arthritis, inflammation was inhibited using an anti-IL-8 antibody [12] (see below). Furthermore, neutrophil influx following injection of MSU crystals into a subcutaneous air pouch was found to be attenuated in mice deficient in the IL-8 receptor CXCR2 [13].
Amplification
Native uncoated MSU crystals can activate leucocytes, but may also be directly membranolytic [14]. In addition, a number of interstitial fluid proteins bind MSU crystals, including immunoglobulins (IgG and IgM), adhesion proteins (e.g. fibronectin) and complement proteins, resulting in protection from lysis, the activation of inflammatory cascades and direct interactions with specific cell surface receptors [15]. A recent report indicates that Toll-like receptors-2 and -4 may be involved in triggering leucocyte activation, although whether these function directly as receptors for MSU crystals remains to be determined [16]. In turn, these interactions promote the release of soluble cellular products which further amplify the inflammatory response, leading to both local arthritis and a systemic acute phase response.
Complement
Activity of the complement system is usually very low in normal uninflamed synovial fluid, but is greatly increased in inflamed synovial fluid obtained from patients with acute gout [17]. MSU crystals activate both the classical and the alternative complement pathway in vitro [18, 19]. A number of complement components, including C1q, C1r and C1s, have been shown to bind MSU crystals [15]. Although MSU crystals also avidly bind IgG, activation of the classical pathway does not require the presence of immunoglobulin, suggesting that MSU crystals directly activate the classical pathway [20]. Activation of the complement pathways leads to the elaboration of C3a and C5a, which act as leucocyte chemoattractants. Furthermore, the coating of MSU crystals with iC3b provides a ligand for interactions with leucocytes (see below). Interestingly, activation of the terminal membrane attack complex of complement appears to play a major role in the generation of IL-8 in response to MSU crystals, and thus neutrophil recruitment into the acutely inflamed joint in gout [21].
Kininogen
Formation of the vasoactive peptide bradykinin may also contribute to amplification of the inflammatory response to MSU crystals. In addition to immunoglobulins and complement components, urate crystals bind plasma kininogen [15]. Bradykinin is formed by the interaction with high molecular weight kininogen on urate crystals, and factor XII and prekallikrein. Bradykinin is able to activate endothelial cells and promote vasodilatation, vascular permeability and arachidonic acid metabolism. In addition, this peptide stimulates sensory nerve endings to induce pain (reviewed in [22]). The relevance of this system to the pathogenesis of the acute gout attack has been emphasized by studies of Brown Norway rats that are devoid of high molecular weight kininogen and poor in kallikrein. In these rats, the inflammatory response to MSU crystals is much reduced [23]. Similarly, bradykinin antagonists also suppress the inflammatory response to urate crystals in vivo [24].
Cellular amplification
MSU crystals can interact with cells by phagocytosis or direct interaction with cell surface receptors. Phagocytosis of MSU crystals is greatly promoted by opsonization by IgG or complement components. Intense infiltration of neutrophils into both synovial membrane and fluid is the hall-mark of acute gout, and these cells provide the main cellular mechanism of inflammatory amplification. In the dog, MSU crystal-induced synovitis can be inhibited by depletion of neutrophils, and this can be reversed by neutrophil reconstitution [25]. A number of neutrophil surface receptors are probably involved in mediating responses to MSU crystals [26, 27], and these include CR3 (CD11b/CD18) and FcγRIII (CD16), which bind crystal-bound iC3b and IgG, respectively [28]. The consequences of neutrophil interaction with MSU crystals include the synthesis and release of a large variety of mediators that promote the vasodilatation, erythema and pain associated with the acute gout attack. These include reactive oxygen species such as superoxide, hydrogen peroxide and singlet oxygen, nitric oxide, leukotriene B4, prostaglandin (PG) E2, anti-microbial peptides, enzymes, IL-1, and chemokines such as S100A8, S100A9 and IL-8 [29–35].
Following phagocytosis of MSU crystals, monocytes also become activated, resulting in expression of a number of proinflammatory genes, including IL-1, TNF-α, IL-6, IL-8 and cyclooxygenase-2 [36–40].
Although the generation of acute inflammatory mediators is usually associated with infiltrating leucocytes, resident stromal cells may also contribute mediators to the response. Thus, synovial fibroblasts can phagocytose MSU crystals and respond by releasing arachidonic acid metabolites such as PGE2 [41].
Recent work has clarified the signalling pathways involved in cellular activation in response to MSU crystals. MSU crystal-induced activation of Src family tyrosine kinases, Syk tyrosine kinase and the ERK1/2, p38 and JNK mitogen-activated protein kinases (MAPK) regulates cellular responses during the acute gout attack [40, 42–45]. Within mononuclear phagocytes, the ERK1/2 MAPK pathway plays a particular role in activator protein-1 (AP-1) and nuclear factor-κB (NF-κB) activation, and subsequent production of proinflammatory mediators following stimulation with MSU crystals [45, 46].
Drugs currently used for the treatment of the acute gout attack inhibit amplification of the inflammatory response to MSU crystals. For example, colchicine, a drug with specific clinical efficacy in acute gout [47], inhibits neutrophil recruitment and activation [32, 33, 48, 49]. Non-steroidal anti-inflammatory agents (NSAIDs) prevent release of PGE2 and other arachidonic acid metabolites from various cells in response to MSU crystals [41, 50]. Through targeting of other pathways involved in the initiation and amplification phases, novel treatments may be identified to prevent or treat the acute gout attack.
Pain in the acute gout attack
A hallmark of the acute crystal arthropathies is severe joint pain. This pain may be due to a number of factors, including local production of prostaglandins and bradykinin, and sensitization of nociceptors [51]. When unmyelinated nerve fibres are stimulated, there is release of neuropeptides such as substance P. Substance P results in vasodilatation, plasma extravasation, leucocyte recruitment, mast cell degranulation, and release of PGs and cytokines. Following intra-articular injection of MSU crystals into domestic chick ankle joints, there is rapid depletion of substance P from peripheral nerves in the synovial and subsynovial tissue [52]. These data implicate substance P as a potential mediator of pain and inflammation in acute gout.
Termination of the acute attack and maintenance of asymptomatic hyperuricaemia
Even in the absence of treatment, the acute inflammatory response in gout is typically self-limiting over 7–10 days. Furthermore, it is well recognized that MSU crystals can be found in the asymptomatic joints of patients with hyperuricaemia [53–55]. These observations imply a balance between the factors within the joint that maintain the non-inflamed state in the presence of MSU crystals and the proinflammatory response that accompanies an acute gout attack.
Stages of the inflammatory response in skin
Injection of MSU crystals into human skin leads to an erythematous reaction that is maximal at 24 h and then spontaneously subsides [56]. A similar response is demonstrated in pig skin, and this model has been used to study the kinetics of endothelial activation and leucocyte trafficking in response to MSU crystals [57]. In this model, the inflammatory response involves several distinct phases (Fig. 1). First, endothelial E-selectin expression increases over 2–6 h, closely paralleled by accumulation of neutrophils and mononuclear phagocytes, and albumin leakage. Secondly, leucocyte accumulation rapidly declines despite persisting E-selectin expression. Thirdly, after peaking 8 h after crystal injection, E-selectin expression falls despite increasing erythema and induration. Finally, the clinical manifestations of inflammation subside despite the continued presence of MSU crystals in the tissue. These observations suggest that down-regulation of the acute inflammatory response may start with the suppression of further leucocyte recruitment by the autoregulation of endothelial activation, possibly through endogenous mechanisms of inactivating NF-κB and other transcription factors that mediate the response to TNF-α and IL-1. However, further mechanisms of down-regulation must exist to account for the gradual loss of erythema after 24 h.
![Kinetics of endothelial E-selectin expression, entry of neutrophils and mononuclear cells into the tissues, and erythema in pig skin after injection of MSU crystals. Reproduced from Haskard and Landis [81] with permission from Biomed Central Ltd.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/rheumatology/44/9/10.1093_rheumatology_keh640/3/m_keh640f1.jpeg?Expires=1716317575&Signature=kG6iBM7Nbn5rHnQKBHrbRR-pi-jh2yuruRwhGaiWn8JCRpY1zW3y42Hjpsh~j88YoBsby-tCTJzOHUZCJVoxUh-NPg0hwHFsNMRLsFqGGp0YQPWgvhL9B5rIm0UOpCeQN-2bCm4C1uuRXzZngRzL8LSZar0kR1yvNLIAYnJkvAWDoSYxW-lDSGINCcsK4A6BrPA5BCNZNLRNts31S-UwyRMAgKiEHieQrK-G9Hts5wbyBv6~UajjLVCqtl7YWorWx1KFv45oNoo9EfGMTnKLrp01ECo52GDB8KquJxuFVQJMNGUdzRbcEaB7jDK7Do~ePy~yjZa9QZeEuWoKnBRm-A__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Kinetics of endothelial E-selectin expression, entry of neutrophils and mononuclear cells into the tissues, and erythema in pig skin after injection of MSU crystals. Reproduced from Haskard and Landis [81] with permission from Biomed Central Ltd.
Mediators of the resolution phase in acute gout
Changes in proteins that coat MSU crystals
Changes in the interstitial fluid proteins that coat MSU crystals may modify inflammation. This effect is at least partly due to lipoproteins containing apolipoproteins B and E [58, 59]. Apolipoprotein E is produced by macrophages and is enriched in inflammatory synovial fluid. Coating of MSU crystals by apolipoprotein E inhibits neutrophil granule release and has been demonstrated in patients with gout [59]. The physiological importance of protein coating has also been suggested by serial sampling of synovial fluid crystals from patients with acute gout, demonstrating that as inflammation subsides MSU crystals become coated with apolipoprotein B. Similar coating has been demonstrated on MSU crystals in resolving air-pouch inflammation [60]. Thus, displacement of proinflammatory IgG by apolipoproteins coating MSU crystals may contribute to resolution of the acute gout attack.
Melanocortins
Products of the hypothalamic-pituitary axis may also influence the resolution of the acute gout attack. The anti-inflammatory properties of the melanocortins, such as adrenocorticotrophic hormone 1-39 (ACTH1-39) and α-melanocyte-stimulating hormone (α-MSH), have been demonstrated in many experimental models. The effects of melanocortin peptides occur through signalling via the five melanocortin receptors (MC-R). The MC3-R has been demonstrated in a murine model of MSU crystal-induced peritonitis, and on macrophages isolated from rat knee joints. ACTH has an anti-inflammatory effect on MSU-induced arthritis in the rat knee joint at a concentration that does not increase circulating corticosteroids. ACTH1-39 is also effective in adrenalectomized rats, confirming that this effect is not due to stimulation of adrenal corticosteroids. This anti-inflammatory action is dependent on signalling through the MC3-R, as the ACTH1-39 effect can be blocked by a selective MC3-R antagonist and can be reproduced by a MC3-R agonist [61, 62].
Peroxisome proliferator-activated receptor γ
Spontaneous resolution of the acute attack may also involve induction of specific intracellular anti-inflammatory pathways. One such example is the induction of peroxisome proliferator-activated receptor γ (PPAR-γ), a member of the nuclear hormone receptor superfamily. This transcription regulator is expressed in a wide variety of cells, including mononuclear phagocytes, and may function as an important negative regulator of the inflammatory response. PPAR-γ ligands inhibit transcription of many proinflammatory genes, including TNF-α, IL-1 and IL-6, cyclooxygenase-2, inducible nitric oxide synthase and matrix metalloproteases (MMPs). The clinical relevance of PPAR-γ in the resolution of acute gout episodes is suggested by experiments demonstrating that MSU crystals induce expression of PPAR-γ mRNA in human monocytes. PPAR-γ can be detected in monocytes by immunohistochemistry 12 h after exposure to MSU crystals. A natural ligand of PPAR-γ, 15-deoxy-PGJ2, is capable of inhibiting the production of TNF-α and IL-1β by MSU-stimulated monocytes, and also inhibits early cellular infiltration in the air pouch model [63].
PPAR-γ ligands also promote monocyte expression of CD36, a scavenger receptor for apoptotic cells [63]. Apoptotic neutrophils are rapidly and efficiently phagocytosed by tissue macrophages. This may protect tissues from damage due to autolysis and spillage of toxic neutrophil contents. Phagocytosis of apoptotic neutrophils by macrophages also promotes the production of anti-inflammatory factors, including transforming growth factor (TGF)-β [64]. Evidence for uptake of such apoptotic cells by macrophages in gout exists in the form of the Reiter cell, which is found in synovial fluid during acute gout attacks [65].
The role of monocyte-macrophage differentiation
Several observations imply that differentiated macrophages may play an important role in the resolution of the acute gout attack. MSU crystals found in asymptomatic joints of patients with hyperuricaemia and interval gout are usually present within macrophages and almost never within neutrophils [66]. This suggests that macrophages can interact with MSU crystals within the joint without triggering an inflammatory response. Within air pouches injected with MSU crystals, macrophage accumulation continues even after 48 h, when neutrophil infiltration has resolved [67]. Moreover, the inhibitory effect of apoptotic neutrophils described above provides evidence that certain phagocytic stimuli can induce an anti-inflammatory macrophage response.
The effect of differentiation upon the response of monocyte-macrophages to MSU crystals has been studied using a panel of mouse cell lines fixed at different stages of maturation (defined by expression of the markers F4/80 and BM 8) [68]. There was close correlation between the level of expression of the surface markers and the capacity to phagocytose latex beads or MSU crystals. However, TNF-α production was not linked to phagocytic activity, since cell lines that were at an intermediate level of maturation expressed the highest concentrations of TNF-α. In contrast, the most mature macrophage lines (MH-S and IC-21) failed to produce TNF-α despite efficient phagocytosis of MSU crystals. Following exposure to MSU crystals, culture supernatants from partially differentiated macrophages, but not from the fully differentiated cell lines, stimulated endothelial activation. Zymosan, an alternative phagocytic stimulus, led to TNF-α production by IC-21 cells, indicating particle specificity of the response. Furthermore, in co-culture experiments, the release of TNF-α by IC-21 cells in response to zymosan was inhibited by a soluble factor released in response to MSU crystals, suggesting that the response to MSU crystals was actively anti-inflammatory rather than neutral.
These experiments using mouse cell lines have now been extended to a model in which human monocytes are differentiated in vitro to macrophages in the presence of autologous serum [69]. Following exposure to MSU crystals, undifferentiated peripheral blood monocytes secreted the cytokines TNF-α, IL-1β and IL-6, induced endothelial activation and promoted neutrophil recruitment under shear flow. However, differentiation of monocytes into mature macrophages over 5 days led to the loss of the capacity to release proinflammatory cytokines capable of activating endothelial cell adhesion molecule expression. As with IC-21 cells, this attenuated response to MSU crystals was particle-specific, zymosan stimulating macrophages to elicit a proinflammatory response. Again, co-culture of MSU crystals with zymosan inhibited zymosan-induced TNF-α production by differentiated macrophages but not immature monocytes (Fig. 2).
![Suppression of zymosan-induced TNF-α synthesis by co-incubation with MSU crystals. Human monocyte/macrophage cultures were incubated with zymosan alone (0.4 mg/ml), MSU crystals alone (0.5 mg/ml), or both stimuli together. TNF-α secretion in undifferentiated monocytes was unaffected by co-mixing of stimuli, but zymosan-induced TNF-α levels in macrophages were significantly inhibited by the addition of MSU crystals. Bars show the mean and s.e.m. *P<0.01. Med, medium; zym, zymosan; Mo, monocyte; Mø, macrophage. Reproduced from Landis et al. [69] with permission from John Wiley & Sons, Inc. Arthritis & Rheumatism © Copyright 2002 the American College of Rheumatology.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/rheumatology/44/9/10.1093_rheumatology_keh640/3/m_keh640f2.jpeg?Expires=1716317575&Signature=3MlhE06aokQ-cSUl7h6NtgTn9qCDG31Q8aplJJe0PQJsaCakoNC9CggmlmTJcaLUekZnNEmiZHs1-UAaUcq5u5mbmR4-mbuJqmutg4FHQcwKnG8-Ebhke66v2E2KhFh4Q2XGzRDsb5yyA0PITPu2Uq~NvwUozJge9CubtQq798PlVGu9WkHl6eNsF0LGhkpfbgoZyQzLMywYbcjQQio15wXV91Jb6oOB6To2D~YScZShZrgAQUnS1yXbZTO-JrqYUgbFgFDeC1jKQ3su2Koja2SPtrAP1MrU5FIPEcmLvbEf2IHBjPJSR1XR2Yjj4WgNxOf2ADGjxAb5UgOQHyOptQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Suppression of zymosan-induced TNF-α synthesis by co-incubation with MSU crystals. Human monocyte/macrophage cultures were incubated with zymosan alone (0.4 mg/ml), MSU crystals alone (0.5 mg/ml), or both stimuli together. TNF-α secretion in undifferentiated monocytes was unaffected by co-mixing of stimuli, but zymosan-induced TNF-α levels in macrophages were significantly inhibited by the addition of MSU crystals. Bars show the mean and s.e.m. *P<0.01. Med, medium; zym, zymosan; Mo, monocyte; Mø, macrophage. Reproduced from Landis et al. [69] with permission from John Wiley & Sons, Inc. Arthritis & Rheumatism © Copyright 2002 the American College of Rheumatology.
Macrophage production of anti-inflammatory cytokines may contribute to resolution of the acute attack. Overexpression of IL-10 using retrovirally transfected IL-10 inhibits macrophage production of TNF-α, macrophage inflammatory protein (MIP)-1α and MIP-1β in vitro, and MSU crystal-induced cellular infiltration in the murine air pouch model in vivo [70]. Thus, IL-10 has the ability to suppress the acute inflammatory response to MSU crystals. However, in vitro differentiated human macrophages do not express significant IL-10 on exposure to MSU crystals, suggesting that IL-10 may not play a major role in suppression of the inflammatory response in this model [71].
High levels of TGF-β1 have been demonstrated in the synovial fluid of patients with acute gout [72], and administration of TGF-β1 significantly inhibited leucocyte infiltration into air pouches injected with MSU crystals [73]. Further studies have now identified TGF-β1 as a key soluble factor in the suppression of MSU-induced inflammation by differentiated macrophages [71]. As monocytes differentiated in vitro towards a macrophage end-point, the loss of the capacity to secrete proinflammatory cytokines in response to MSU crystals was paralleled by a gain in the capacity to release TGF-β1. Functional effects of TGF-β1 in this model system include the suppression of (i) monocyte proinflammatory cytokine release in response to MSU crystals, (ii) endothelial cell activation in response to monocyte-derived cytokines, and (iii) macrophage release of TNF-α in response to zymosan. However, not all effects of TGF-β are suppressive and this growth factor may contribute to fibroblast proliferation and the physical encasing of crystals away from contact with leucocytes. Certainly, synovial tissue taken from patients with acute gout demonstrates marked fibroblast proliferation within the lining layer.
Taken together, these data support a role for monocyte differentiation in the resolution of acute inflammation in gout. As with any in vitro model, the interpretation of the data needs to be qualified by the possibility that macrophage differentiation in vitro may not faithfully reproduce the in vivo situation. In this respect, it is reassuring that monocytes and macrophages derived from a skin blister model showed the same disparity in cytokine secretion in response to MSU crystals as in vitro differentiated cells, the macrophage end-point appearing rather earlier in vivo (40 h as opposed to 5 days), consistent with the kinetics of a typical attack of gout (Fig. 3) [71].
![MSU crystal induction of TNF-α and TGF-β1 in human skin blister leucocytes. Cantharidin skin blisters were initiated by topical application of the vesicant cantharidin to the forearm of normal volunteers. Blister exudate cells were collected and pooled at the 16- and 40-h time points, corresponding to the acute and resolving phases respectively. Secretion of TNF-α and TGF-β1 were measured following culture for 24 h in the presence and absence of MSU crystals (0.5 mg/ml). Values show the mean and s.d. from three donors. Reproduced from Yagnik et al. [71] with permission from John Wiley & Sons, Inc. Arthritis & Rheumatism © Copyright 2004 the American College of Rheumatology.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/rheumatology/44/9/10.1093_rheumatology_keh640/3/m_keh640f3.jpeg?Expires=1716317575&Signature=Je4EZChVUul3GtbHteGHYUrrLe3Zd25hO0qARCpXc-CZcFdb1BqP5YQckYJjGMOVutPo7KKY~r7ATkaNUwngoz4ATowZy4jpl4U-PjfykOmIEU1IUSZ4f~c6l9cH6empse4Ye79Z~QT5Aehlcn3MMrW4B~98T1~z-Ec7dx0LqPynnL9f9WuX3Xi~OTp5~7yhyGSy0iDINI9komeAagih~Clretz3zy8qJVh~6N1OmKdqgFryj-HM6sWW9pX6w38J8A7Di2ZUmL1gPfBL9R2~MESX3-qaERJ1aS~LEOU76tHUnAC9N4n2j3l-QzwkM4B1EgpMIoQdRDRDxliVMmMYXw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
MSU crystal induction of TNF-α and TGF-β1 in human skin blister leucocytes. Cantharidin skin blisters were initiated by topical application of the vesicant cantharidin to the forearm of normal volunteers. Blister exudate cells were collected and pooled at the 16- and 40-h time points, corresponding to the acute and resolving phases respectively. Secretion of TNF-α and TGF-β1 were measured following culture for 24 h in the presence and absence of MSU crystals (0.5 mg/ml). Values show the mean and s.d. from three donors. Reproduced from Yagnik et al. [71] with permission from John Wiley & Sons, Inc. Arthritis & Rheumatism © Copyright 2004 the American College of Rheumatology.
Tophaceous gout
It is well established that only a proportion of hyperuricaemic individuals develop gout [74]. Given that MSU crystals can be found within synovial mononuclear cells in asymptomatic joints of hyperuricaemic individuals [66], it seems possible that resident tissue macrophages may play a key role in maintaining the asymptomatic state in hyperuricaemia by clearing crystals as and when they form. In some individuals, persistent hyperuricaemia leads to the formation of tophaceous deposits of MSU crystals, typically in subcutaneous and periarticular areas. The reasons why some individuals are susceptible to the development of tophi is not known, but could include the formation of crystals at a rate that exceeds the handling capacity of tissue macrophages or possibly the failure of macrophages to differentiate to an end-point which does not show a proinflammatory response to crystal uptake.
Microscopically, tophi are granulomas of mono- and multinucleated macrophages surrounding a core of debris and MSU crystals, encased by dense connective tissue [75]. The gradation of size and urate content of gouty tophi suggests a progressive enlargement and maturation [75]. Within the tophus, macrophages express mature, late differentiation markers and show high levels of apoptosis. However, in associated perivascular regions, there is a predominance of mononucleated monocyte-macrophages expressing surface markers of recent migration [76]. These data suggest that the development of the gouty tophus is a dynamic process with a low level continuous recruitment, proinflammatory activation, maturation and turnover of monocyte-macrophages. This view is supported by the detection of TNF-α in tophaceous tissue [37, 76].
Tophi are frequently associated with tissue destruction of cartilage and bone. Several factors have been identified within tophaceous material that may contribute to such erosive disease. Monocyte-macrophages within the gouty tophus produce gelatinase A (MMP-2) and gelatinase B (MMP-9) [76]. These enzymes are capable of degrading type IV and type V collagen, elastin and gelatin. MMP-9 expression is induced in macrophages by MSU crystals in vitro in a dose-dependent manner [77]. Resident stromal cells also produce MMPs on exposure to MSU crystals, synovial fibroblasts producing collagenase (MMP-1) [78, 79] and chondrocytes producing stromelysin 1 (MMP-3) [80]. Release of such enzymes may play a role in degradation of matrix in articular structures adjacent to the tophus.
Conclusion
The inflammatory response in gout is characterized by initiation of the acute attack, leucocyte recruitment, amplification and subsequent resolution. These clinical manifestations of disease are due to complex interactions between various cell types, including mast cells, endothelial cells, neutrophils, macrophages and synovial fibroblasts. It is possible that the balance of monocytes and differentiated macrophages plays a key role in modulating the inflammatory response to MSU crystals. A hypothetical model of the role of monocyte-macrophage differentiation in gout is shown in Fig. 4. Further analysis of the pathways that regulate the cellular response to inflammatory microcrystals may identify potential therapeutic targets for the management of gout.
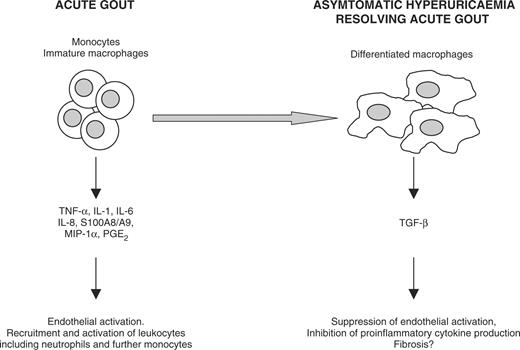
A model of the differential roles of monocytes and macrophages in the inflammatory response to MSU crystals. The model proposes that monocytes and immature macrophages act to stimulate and amplify an acute attack of gout, whereas differentiated macrophages may play an anti-inflammatory role in terminating an acute attack and in preserving the asymptomatic state through production of TGF-β.
The authors have declared no conflicts of interest.
References
Agudelo CA, Schumacher HR. The synovitis of acute gouty arthritis. A light and electron microscopic study.
Schumacher HR. Pathology of the synovial membrane in gout. Light and electron microscopic studies. Interpretation of crystals in electron micrographs.
Tak HK, Cooper SM, Wilcox WR. Studies on the nucleation of monosodium urate at 37 degrees C.
McGill NW, Dieppe PA. Evidence for a promoter of urate crystal formation in gouty synovial fluid.
Perl-Treves D, Addadi L. A structural approach to pathological crystallizations. Gout: the possible role of albumin in sodium urate crystallization.
Getting SJ, Flower RJ, Parente L et al. Molecular determinants of monosodium urate crystal-induced murine peritonitis: a role for endogenous mast cells and a distinct requirement for endothelial-derived selectins.
Meng H, Tonnesen MG, Marchese MJ, Clark RA, Bahou WF, Gruber BL. Mast cells are potent regulators of endothelial cell adhesion molecule ICAM-1 and VCAM-1 expression.
Chapman PT, Yarwood H, Harrison AA et al. Endothelial activation in monosodium urate monohydrate crystal-induced inflammation: in vitro and in vivo studies on the roles of tumor necrosis factor alpha and interleukin-1.
Russell IJ, Mansen C, Kolb LM, Kolb WP. Activation of the fifth component of human complement (C5) induced by monosodium urate crystals: C5 convertase assembly on the crystal surface.
Ryckman C, McColl SR, Vandal K et al. Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis.
Nishimura A, Akahoshi T, Takahashi M et al. Attenuation of monosodium urate crystal-induced arthritis in rabbits by a neutralizing antibody against interleukin-8.
Terkeltaub R, Baird S, Sears P, Santiago R, Boisvert W. The murine homolog of the interleukin-8 receptor CXCR-2 is essential for the occurrence of neutrophilic inflammation in the air pouch model of acute urate crystal-induced gouty synovitis.
Kozin F, Ginsberg MH, Skosey JL. Polymorphonuclear leukocyte responses to monosodium urate crystals: modification by adsorbed serum proteins.
Terkeltaub R, Tenner AJ, Kozin F, Ginsberg MH. Plasma protein binding by monosodium urate crystals. Analysis by two-dimensional gel electrophoresis.
Liu-Bryan R, Rose DM, Sydlaske A, Terkeltaub R. Engagement of toll-like receptor 2 is the central trigger of acute neutrophilic gouty inflammation in vivo.
Hasselbacher P. C3 activation by monosodium urate monohydrate and other crystalline material.
Doherty M, Whicher JT, Dieppe PA. Activation of the alternative pathway of complement by monosodium urate monohydrate crystals and other inflammatory particles.
Giclas PC, Ginsberg MH, Cooper NR. Immunoglobulin G independent activation of the classical complement pathway by monosodium urate crystals.
Tramontini N, Huber C, Liu-Bryan R, Terkeltaub R, Kilgore KS. Central role of complement membrane attack complex in monosodium urate crystal-induced neutrophilic rabbit knee synovitis.
Kaplan AP, Joseph K, Silverberg M. Pathways for bradykinin formation and inflammatory disease.
Damas J, Remacle-Volon G, Adam A. The role of the kinin system in various inflammatory models in the rat.
Damas J, Remacle-Volon G. Influence of a long-acting bradykinin antagonist, Hoe 140, on some acute inflammatory reactions in the rat.
Phelps P, McCarty DJ Jr. Crystal-induced inflammation in canine joints. II. Importance of polymorphonuclear leukocytes.
Terkeltaub RA, Sklar LA, Mueller H. Neutrophil activation by inflammatory microcrystals of monosodium urate monohydrate utilizes pertussis toxin-insensitive and -sensitive pathways.
Onello E, Traynor-Kaplan A, Sklar L, Terkeltaub R. Mechanism of neutrophil activation by an unopsonized inflammatory particulate. Monosodium urate crystals induce pertussis toxin-insensitive hydrolysis of phosphatidylinositol 4,5-bisphosphate.
Barabe F, Gilbert C, Liao N, Bourgoin SG, Naccache PH. Crystal-induced neutrophil activation VI. Involvement of FcgammaRIIIB (CD16) and CD11b in response to inflammatory microcrystals.
Abramson S, Hoffstein ST, Weissmann G. Superoxide anion generation by human neutrophils exposed to monosodium urate.
Simchowitz L, Atkinson JP, Spilberg I. Stimulation of the respiratory burst in human neutrophils by crystal phagocytosis.
Gilbert C, Poubelle PE, Borgeat P, Pouliot M, Naccache PH. Crystal-induced neutrophil activation: VIII. Immediate production of prostaglandin E2 mediated by constitutive cyclooxygenase 2 in human neutrophils stimulated by urate crystals.
Roberge CJ, Grassi J, De Medicis R et al. Crystal-neutrophil interactions lead to interleukin-1 synthesis.
Roberge CJ, de Medicis R, Dayer JM, Rola-Pleszczynski M, Naccache PH, Poubelle PE. Crystal-induced neutrophil activation. V. Differential production of biologically active IL-1 and IL-1 receptor antagonist.
Hachicha M, Naccache PH, McColl SR. Inflammatory microcrystals differentially regulate the secretion of macrophage inflammatory protein 1 and interleukin 8 by human neutrophils: a possible mechanism of neutrophil recruitment to sites of inflammation in synovitis.
Ryckman C, Gilbert C, De Medicis R, Lussier A, Vandal K, Tessier PA. Monosodium urate monohydrate crystals induce the release of the proinflammatory protein S100A8/A9 from neutrophils.
di Giovine FS, Malawista SE, Nuki G, Duff GW. Interleukin 1 (IL 1) as a mediator of crystal arthritis. Stimulation of T cell and synovial fibroblast mitogenesis by urate crystal-induced IL 1.
di Giovine FS, Malawista SE, Thornton E, Duff GW. Urate crystals stimulate production of tumor necrosis factor alpha from human blood monocytes and synovial cells. Cytokine mRNA and protein kinetics, and cellular distribution.
Guerne PA, Terkeltaub R, Zuraw B, Lotz M. Inflammatory microcrystals stimulate interleukin-6 production and secretion by human monocytes and synoviocytes.
Terkeltaub R, Zachariae C, Santoro D, Martin J, Peveri P, Matsushima K. Monocyte-derived neutrophil chemotactic factor/interleukin-8 is a potential mediator of crystal-induced inflammation.
Pouliot M, James MJ, McColl SR, Naccache PH, Cleland LG. Monosodium urate microcrystals induce cyclooxygenase-2 in human monocytes.
Wigley FM, Fine IT, Newcombe DS. The role of the human synovial fibroblast in monosodium urate crystal-induced synovitis.
Liu R, Aupperle K, Terkeltaub R. Src family protein tyrosine kinase signaling mediates monosodium urate crystal-induced IL-8 expression by monocytic THP-1 cells.
Gaudry M, Gilbert C, Barabe F, Poubelle PE, Naccache PH. Activation of Lyn is a common element of the stimulation of human neutrophils by soluble and particulate agonists.
Desaulniers P, Fernandes M, Gilbert C, Bourgoin SG, Naccache PH. Crystal-induced neutrophil activation. VII. Involvement of Syk in the responses to monosodium urate crystals.
Liu R, O'Connell M, Johnson K, Pritzker K, Mackman N, Terkeltaub R. Extracellular signal-regulated kinase 1/extracellular signal-regulated kinase 2 mitogen-activated protein kinase signaling and activation of activator protein 1 and nuclear factor kappaB transcription factors play central roles in interleukin-8 expression stimulated by monosodium urate monohydrate and calcium pyrophosphate crystals in monocytic cells.
Jaramillo M, Godbout M, Naccache PH, Olivier M. Signaling events involved in macrophage chemokine expression in response to monosodium urate crystals.
Ahern MJ, Reid C, Gordon TP, McCredie M, Brooks PM, Jones M. Does colchicine work? The results of the first controlled study in acute gout.
Phelps P. Polymorphonuclear leukocyte motility in vitro. IV. Colchicine inhibition of chemotactic activity formation after phagocytosis of urate crystals.
Roberge CJ, Gaudry M, de Medicis R, Lussier A, Poubelle PE, Naccache PH. Crystal-induced neutrophil activation. IV. Specific inhibition of tyrosine phosphorylation by colchicine.
Gordon TP, Kowanko IC, James M, Roberts-Thomson PJ. Monosodium urate crystal-induced prostaglandin synthesis in the rat subcutaneous air pouch.
Gentle MJ. Sodium urate arthritis: effects on the sensory properties of articular afferents in the chicken.
Lunam CA, Gentle MJ. Substance P immunoreactive nerve fibres in the domestic chick ankle joint before and after acute urate arthritis.
Gordon TP, Bertouch JV, Walsh BR, Brooks PM. Monosodium urate crystals in asymptomatic knee joints.
Pascual E. Persistence of monosodium urate crystals and low-grade inflammation in the synovial fluid of patients with untreated gout.
Pascual E, Batlle-Gualda E, Martinez A, Rosas J, Vela P. Synovial fluid analysis for diagnosis of intercritical gout.
Dieppe PA, Doherty M, Papadimitriou GM. Inflammatory responses to intradermal crystals in healthy volunteers and patients with rheumatic diseases.
Chapman PT, Jamar F, Harrison AA et al. Characterization of E-selectin expression, leucocyte traffic and clinical sequelae in urate crystal-induced inflammation: an insight into gout.
Terkeltaub R, Curtiss LK, Tenner AJ, Ginsberg MH. Lipoproteins containing apoprotein B are a major regulator of neutrophil responses to monosodium urate crystals.
Terkeltaub RA, Dyer CA, Martin J, Curtiss LK. Apolipoprotein (apo) E inhibits the capacity of monosodium urate crystals to stimulate neutrophils. Characterization of intraarticular apo E and demonstration of apo E binding to urate crystals in vivo.
Ortiz-Bravo E, Sieck MS, Schumacher HR Jr. Changes in the proteins coating monosodium urate crystals during active and subsiding inflammation. Immunogold studies of synovial fluid from patients with gout and of fluid obtained using the rat subcutaneous air pouch model.
Getting SJ, Allcock GH, Flower R, Perretti M. Natural and synthetic agonists of the melanocortin receptor type 3 possess anti-inflammatory properties.
Getting SJ, Christian HC, Flower RJ, Perretti M. Activation of melanocortin type 3 receptor as a molecular mechanism for adrenocorticotropic hormone efficacy in gouty arthritis.
Akahoshi T, Namai R, Murakami Y et al. Rapid induction of peroxisome proliferator-activated receptor gamma expression in human monocytes by monosodium urate monohydrate crystals.
Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF.
Selvi E, Manganelli S, De Stefano R, Frati E, Marcolongo R. CD36 and CD14 immunoreactivity of Reiter cells in inflammatory synovial fluids.
Pascual E, Jovani V. A quantitative study of the phagocytosis of urate crystals in the synovial fluid of asymptomatic joints of patients with gout.
Schiltz C, Liote F, Prudhommeaux F et al. Monosodium urate monohydrate crystal-induced inflammation in vivo: quantitative histomorphometric analysis of cellular events.
Yagnik DR, Hillyer P, Marshall D et al. Noninflammatory phagocytosis of monosodium urate monohydrate crystals by mouse macrophages. Implications for the control of joint inflammation in gout.
Landis RC, Yagnik DR, Florey O et al. Safe disposal of inflammatory monosodium urate monohydrate crystals by differentiated macrophages.
Murakami Y, Akahoshi T, Kawai S, Inoue M, Kitasato H. Antiinflammatory effect of retrovirally transfected interleukin-10 on monosodium urate monohydrate crystal-induced acute inflammation in murine air pouches.
Yagnik DR, Evans BJ, Florey O, Mason JC, Landis RC, Haskard DO. Macrophage release of transforming growth factor beta1 during resolution of monosodium urate monohydrate crystal-induced inflammation.
Fava R, Olsen N, Keski-Oja J, Moses H, Pincus T. Active and latent forms of transforming growth factor beta activity in synovial effusions.
Liote F, Prudhommeaux F, Schiltz C et al. Inhibition and prevention of monosodium urate monohydrate crystal-induced acute inflammation in vivo by transforming growth factor beta1.
Campion EW, Glynn RJ, DeLabry LO. Asymptomatic hyperuricemia. Risks and consequences in the Normative Aging Study.
Palmer DG, Highton J, Hessian PA. Development of the gout tophus. An hypothesis.
Schweyer S, Hemmerlein B, Radzun HJ, Fayyazi A. Continuous recruitment, co-expression of tumour necrosis factor-alpha and matrix metalloproteinases, and apoptosis of macrophages in gout tophi.
Hsieh MS, Ho HC, Chou DT et al. Expression of matrix metalloproteinase-9 (gelatinase B) in gouty arthritis and stimulation of MMP-9 by urate crystals in macrophages.
Hasselbacher P. Stimulation of synovial fibroblasts by calcium oxalate and monosodium urate monohydrate. A mechanism of connective tissue degradation in oxalosis and gout.
McMillan RM, Vater CA, Hasselbacher P, Hahn J, Harris ED Jr. Induction of collagenase and prostaglandin synthesis in synovial fibroblasts treated with monosodium urate crystals.
Liu R, Liote F, Rose DM, Merz D, Terkeltaub R. Proline-rich tyrosine kinase 2 and Src kinase signaling transduce monosodium urate crystal-induced nitric oxide production and matrix metalloproteinase 3 expression in chondrocytes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments