
Abstract
Gut commensal microbes shape the mucosal immune system by regulating the differentiation and expansion of several types of T cell1,2,3,4,5. Clostridia, a dominant class of commensal microbe, can induce colonic regulatory T (Treg) cells, which have a central role in the suppression of inflammatory and allergic responses3. However, the molecular mechanisms by which commensal microbes induce colonic Treg cells have been unclear. Here we show that a large bowel microbial fermentation product, butyrate, induces the differentiation of colonic Treg cells in mice. A comparative NMR-based metabolome analysis suggests that the luminal concentrations of short-chain fatty acids positively correlates with the number of Treg cells in the colon. Among short-chain fatty acids, butyrate induced the differentiation of Treg cells in vitro and in vivo, and ameliorated the development of colitis induced by adoptive transfer of CD4+CD45RBhi T cells in Rag1−/− mice. Treatment of naive T cells under the Treg-cell-polarizing conditions with butyrate enhanced histone H3 acetylation in the promoter and conserved non-coding sequence regions of the Foxp3 locus, suggesting a possible mechanism for how microbial-derived butyrate regulates the differentiation of Treg cells. Our findings provide new insight into the mechanisms by which host–microbe interactions establish immunological homeostasis in the gut.
Similar content being viewed by others


Main
Germ-free mice have an enlarged caecum because of the accumulation of hydrated dietary fibre components. Colonization with chloroform-resistant bacteria (CRB)6 normalizes caecal size, probably through fermentation of these components by the CRB (Fig. 1a, b). Therefore, CRB-derived dietary fibre metabolites could be responsible for the induction of colonic Treg cells. To assess this possibility, CRB-associated mice were fed either a low-fibre diet (LFD) or a high-fibre diet (HFD). In CRB-associated HFD-fed (CRB-HFD) mice, the expression of neuropilin-1− and Helios−(also known as IKZF2−) Treg cells, which represent the peripherally generated Treg cell population7,8,9, was significantly increased (Fig. 1c, d and Supplementary Figs 1 and 2). Consistent with previous observations3, the effect of CRB on Treg cell induction was restricted to the colon and not the mesenteric lymph nodes or spleen (Supplementary Fig. 3). In addition, the expression of CD103+-activated Treg cells was also increased in CRB-HFD mice (Fig. 1d). Notably, a LFD compromised the effects of CRB colonization on Treg cell induction (Fig. 1c, d), despite the comparable number and composition of gut microbes predominantly annotated as Clostridiales (Supplementary Figs 4–6). These observations strongly suggest that metabolic differences in gut microbes are responsible for the differential Treg cell inducibility between CRB-HFD and CRB-LFD mice.

a, b, The caecum size of germ-free (GF) and CRB-associated mice. Representative caecum images are shown in a. Scale bars, 1 cm. c, d, The expression of neuropilin-1 and CD103 in Foxp3+ T cells in germ-free and CRB-associated mice fed a HFD or LFD for 4 weeks. Representative FACS plots gated on CD3ε+CD4+ are shown in c. P value was determined by one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test. e, f, Caecal metabolomic profiling by NMR of germ-free and CRB mice fed a HFD or LFD. The results of PCA on the 1H-NMR data (e) and loading scatter plot (f) are shown. Proportions of the first (PC1) and second (PC2) principal components are 85.1% and 10.3%, respectively. The ellipse denotes the 95% significance limit of the model, as defined by Hotelling’s t-test. g, h, Organic acids in caecal contents (g) and the proximal colonic tissue (h) were measured by gas chromatography–mass spectrometry. Ace, acetate; but, butyrate; pro, propionate. Error bars indicate s.e.m. (b, n = 5) or s.d. (d, n = 10; g, h, n = 7). All data are representative of two (e–h) or three (a–d) independent experiments.
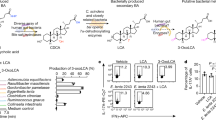
Indeed, principal component analysis (PCA) and orthogonal partial least squares discriminate analysis of the NMR-based metabolome data indicated that the amount of caecal short-chain fatty acids (SCFAs), namely acetate, propionate and butyrate, was higher in CRB-HFD than in CRB-LFD mice (Fig. 1e, f, Supplementary Fig. 7 and Supplementary Table 1). Quantitative analysis confirmed a significant increase in acetate and butyrate in the caecal content and colonic tissues of CRB-HFD mice (Fig. 1g, h). Luminal GABA (γ-aminobutyric acid), l-leucine and l-isoleucine were also increased in CRB-HFD mice (Fig. 1f and Supplementary Fig. 7). To test whether these metabolites can induce Treg cells in vitro, splenic naive (CD44lo CD62Lhi) CD4+ T cells were cultured in the presence of T-cell antigen receptor and CD28 signalling plus TGF-β with or without each metabolite. Butyrate significantly increased the frequency of Foxp3+ cells (Fig. 2a, b). Propionate showed a moderate effect, whereas the other metabolites, including acetate, did not have any effect on Treg cell induction at physiological concentrations (Fig. 2a, b and Supplementary Fig. 8).

a, b, Naive CD4+ T cells were stimulated with immobilized anti-CD3 and soluble anti-CD28 monoclonal antibodies in the absence (untreated; UT) or presence of acetate, propionate or butyrate. Representative FACS plots are shown in a. c, Amount of caecal SCFA in the mice fed with control HAMS diet or diets containing HAMSA, HAMSP or HAMSB (SA, SP or SB, respectively). d, e, FACS profile of colonic lamina propria cells. The representative FACS plots gated on CD3ε+CD4+ cells are shown in d. e, The frequency of neuropilin-1− and neuropilin-1+ Foxp3+cells among CD4+ T cells. f, g, The percentage and total number of OVA-induced Foxp3+ cells in mice fed with HAMS control (Ctrl) or HAMSB (SB). Representative FACS plots gated on CD3ε+CD4+CD45.2+are shown in f. CD45.2+Vα2+CD4+ T cells were sorted and adoptively transferred into CD45.1+ C57BL/6 recipients, followed by administration of drinking water containing 1% ovalbumin (OVA) for 9 days before analysis. h, Percentage of IL-10+ cells among CD25+FR4+ colonic Treg cells in SPF C57BL/6 mice fed with HAMS or HAMSB. Error bars indicate s.d. (b, n = 3; c, n = 4; e, n = 5; g, n = 4 for control and n = 5 for SB; h, n = 5). P values determined by non-parametrical Mann–Whitney U test (g, h) or one-way ANOVA followed by Tukey’s post-hoc test (b, e). All data are representative of at least two independent experiments. NS, not significant.
To corroborate the role of butyrate in Treg cell induction in vivo, specific pathogen-free (SPF) C57BL/6 mice were fed modified diets containing acetylated, propionylated or butyrylated high-amylose maize starches10 (HAMSA, HAMSP or HAMSB, respectively) to increase the luminal levels of the corresponding SCFA (Fig. 2c). Consistent with the in vitro observations, colonic Treg cells were significantly augmented when the diet contained HAMSB, whereas HAMSP slightly and HAMSA barely induced Treg cells in SPF mice (Fig. 2d, e and Supplementary Fig. 9). The HAMSB diet also increased the number of ovalbumin (OVA)-specific colonic OT-II Foxp3+ cells from adoptively transferred OT-II naive CD4+ T cells (Fig. 2f, g). Because butyrate did not affect the survival or the proliferation of colonic Treg cells (Supplementary Figs 10 and 11), this increase probably reflects a role for this SCFA in regulating Treg cell differentiation. Furthermore, the intake of HAMSB increased colonic Treg cells in germ-free mice mono-associated with Bacteroides thetaiotaomicron, an organism devoid of butyrate-producing and Treg-cell-inducing activities per se3 (Supplementary Fig. 12). Under germ-free conditions, HAMSB did not increase Treg cells, suggesting that the presence of commensal bacteria is a prerequisite for Treg cell induction (Supplementary Fig. 13).
Interleukin (IL)-10 production by Treg cells is well documented to be required for containment of inflammatory responses in mucosal tissues including the colon11, and the colonization with a mixture of Clostridiales is sufficient to induce IL-10-producing Treg cells3. Likewise, the intake of HAMSB significantly increased IL-10-producing Treg cells in the colon (Fig. 2h). Collectively, these observations illustrate that butyrate has a key role in the commensal-microbe-mediated differentiation of functional Treg cells in the colon.
We also explored the effect of butyrate on the differentiation of naive T cells into other CD4+ T-cell subsets. Treatment with butyrate or feeding of HAMSB did not affect the induction of T-bet, GATA3 and RORγt, the master regulators of T helper 1 (TH1), TH2 and TH17 cells, respectively (Supplementary Figs 14 and 15). Notably, butyrate promoted the induction of Foxp3+ cells even under TH1- and TH17-polarizing conditions. These data strongly suggest that butyrate preferentially induces Treg cell differentiation.
The requirement for the Toll-like receptor (TLR)–MyD88 signalling pathway12 in commensal microbe-induced Treg cell expansion seems to be variable depending on the type of bacteria involved3,4,5,13. We observed that treatment of SPF Myd88−/− Ticam1−/− mice with HAMSB induced colonic Treg cells (Supplementary Fig. 16) to the same extent as in wild-type mice (Fig. 2d, e). Therefore, TLR–MyD88 signalling is dispensable for butyrate-dependent Treg cell induction in the colon.
Butyrate is well known to regulate gene expression epigenetically by inhibiting histone deacetylases (HDACs)14,15, specifically classes IIa and I of the four HDAC classes identified in mammals. Because class IIa HDAC has been reported to suppress Treg cell expansion16,17, butyrate may influence histone acetylation of gut CD4+ T cells to regulate epigenetically the transcription of the genes responsible for Treg cell induction. To verify this hypothesis, we performed chromatin immunoprecipitation sequencing (ChIP-seq) analysis of naive CD4+ T cells treated with or without butyrate under Treg-cell-polarizing conditions. Butyrate slightly increased genome-wide histone H3 acetylation (Supplementary Fig. 17). Consistently, the histone acetylation status of the promoter regions of most transcription factors was unchanged or moderately upregulated (Fig. 3a–c), and only 70 transcription factors (4.73% of the genes categorized as a transcription factor), including Foxp3, were highly acetylated after butyrate exposure (Fig. 3b, pink area). Importantly, this increased histone acetylation was positively correlated with gene expression (Fig. 3c). Foxp3 expression is regulated not only by its promoter but also by intragenic enhancer elements termed conserved noncoding sequence (CNS) 1–3 (ref. 18). Butyrate upregulated histone H3 acetylation at both the promoter and CNS3 of the Foxp3 gene locus 1 day before Foxp3 induction (Fig. 3a, d). Furthermore, butyrate exposure gradually increased the acetylated histone H3 status of the CNS1 region over the course of Treg cell differentiation. In sharp contrast, no such epigenetic modifications were observed in the Tbx21, Gata3 and Rorc genes after butyrate exposure (Supplementary Fig. 18). CNS1, which contains binding motifs for Smad3, NFAT and retinoic acid receptor, is important for the peripheral induction of Foxp3 (ref. 18). Conversely, CNS3 contains a c-Rel binding motif, which establishes the Foxp3-specific enhanceosome and thus greatly enhances the probability of Foxp3 induction19,20. We detected little, if any, increase in histone H3 acetylation at the c-Rel, NFAT, Smad3 or Stat5 promoters after butyrate exposure (Supplementary Fig. 19). Correspondingly, expression levels of these genes remained unchanged (Supplementary Fig. 20). On the basis of these observations, we propose that butyrate probably augments the accessibility of these transcriptional regulators to the enhancer elements as well as the promoter region through acetylation of the Foxp3 gene locus.

a, Time-course analysis on the differentiation of naive T cells into Foxp3+ Treg cells. Naive CD4+ T cells were cultured under Treg-cell-inducing condition in the absence (−) or presence (+) of butyrate. b–d, Acetylated histone H3 (AcH3) status of transcriptional regulator genes at day 1 after butyrate exposure was determined by ChIP-seq analysis. Distribution of acetylation changes in the gene loci encoding transcription factors (TFs) was fitted using multiple Gaussian distributions with the expectation–maximization algorithm (b). Using this analysis, we identified two groups representing upper (highly acetylated) and lower (moderately or not acetylated) transcriptional regulator genes after butyrate treatment. The threshold was set so that the false discovery ratio (FDR) in the upper group was 0.01, where 99% of transcriptional regulators with a high acetylation changes belonged to the upper group (b, pink area). Correlation of gene expression changes and the acetylated H3 status in transcriptional regulators is shown in c. Gene expression was analysed by microarray at day 2. The normalized number of sequence tags of DNA at the Foxp3 promoter and gene body is shown in d. ChrX, chromosome X. e, ChIP quantitative PCR (qPCR) analysis of the Foxp3 promoter and enhancer regions was performed in CD4+T cells cultured in the presence or absence of butyrate, using anti-AcH3 antibody or rabbit IgG as a negative control. Error bars indicate s.d (a, e, n = 3).
Finally, we investigated the function of the butyrate-induced Treg cells in chronic intestinal inflammation by adoptively transferring CD4+CD45RBhi naive T cells into Rag1−/− mice21. Over the course of the experiment, the recipient mice were fed either a control HAMS- or a HAMSB-containing diet. The HAMS-fed group developed wasting disease in association with severe colitis. However, the intake of HAMSB, but not HAMSA or HAMSP, ameliorated the colitis development (Fig. 4a–f and Supplementary Fig. 21). This beneficial effect is at least partly attributed to the induction of colonic Treg cells by butyrate. In support of this notion, depletion of Treg cells abrogated the protective effect of HAMSB (Fig. 4g–k).

a–f, Experimental colitis was induced by adoptive transfer of CD4+CD45RBhi T cells in Rag1−/− mice fed with HAMS or HAMSB. a, Body weight change was monitored. b–f, The number of colonic lamina propria (LP)-infiltrating mononuclear cells (b) and histological score (c), representative colonic specimens stained with haematoxylin and eosin (HE) and Alcian blue (AB) (d), representative immunofluorescent staining images for CD4, CD11b, CD11c and nuclei (blue) (e) and the frequency of Foxp3+ cells in the colonic mucosa (f) were analysed at 6 weeks after the cell transfer. g–k, CD4+CD45RBhi T cells from Foxp3hCD2 reporter mice were transferred into Rag1−/− recipients, which were injected intravenously with anti-human CD2 monoclonal antibody twice at 4 and 5 weeks after the cell transfer. Depletion of Foxp3+ cells in the colonic lamina propria was confirmed by FACS analysis (g). The mice were analysed for colitis development at 6 weeks after transfer (h–k). Turquoise blue represents goblet cells (d, h, bottom). Error bars indicate s.d. (a, b, c, f, n = 5; i–k, n = 4). P values were determined by two-way repeated measures ANOVA followed by Tukey’s test (a) and non-parametrical Mann–Whitney U test (b, c, i–k). **P < 0.01 (a). Scale bars, 200 μm (d, e, h). All data are representative of two independent experiments.
The present study demonstrates that butyrate produced by gut microbes induces functional colonic Treg cells, specifically among CD4+ T-cell subsets, via T-cell intrinsic epigenetic upregulation of the Foxp3 gene. However, we do not formally exclude the possibility that effects of butyrate other than histone acetylation status, or targets of HDAC other than histones, may also be involved in Treg cell differentiation. SCFAs have also been reported to act through cell surface signalling receptors such as G-protein-coupled receptor 43 (GPR43) to execute some of their functions22; however, this is unlikely to be the case for the effect of butyrate on Treg cells, because acetate, which is a potent GPR43 ligand23, failed to ameliorate T-cell-dependent experimental colitis or induce Treg cell differentiation (Fig. 2a–e and Supplementary Fig. 21). In addition, the expression of GPR43 is restricted to myeloid cells among haematopoietic cell lineages. Nevertheless, we do not exclude the possibility that butyrate may stimulate other GPRs on myeloid cells including dendritic cells to facilitate Treg cell differentiation.
Butyrate has been appreciated for its beneficial effects on the host, including trophic and anti-inflammatory effects on epithelial cells24. The butyrate transporter is downregulated in the colonic mucosa of patients with inflammatory bowel diseases (IBD)25. In addition, butyrate-producing bacteria are decreased in the intestinal microbiota at the gut mucosa and in the faecal samples of patients with IBD compared to control patients26. These facts suggest that butyrate insufficiency may be involved in the pathogenesis of IBD. In support of this idea, butyrate enema, alone or as a cocktail of SCFAs, has been shown to ameliorate colonic inflammation in patient with IBD27,28, although the underlying mechanisms were not fully understood. Our findings not only link butyrate to commensal microbe-mediated induction of functional Treg cells in the colonic mucosa, but also provide molecular insight into the therapeutic application of butyrate and how a metabolite produced by colonic microbial fermentation mediates host–microbial crosstalk for establishment of gut immune homeostasis (Supplementary Fig. 22).
Methods Summary
Animals
Germ-free IQI mice were housed in germ-free isolators in the animal facility of RIKEN. Eight-to-ten-week-old male or female germ-free mice were orally inoculated with 3% (v/v) chloroform-treated murine faecal suspension (CRB)6. Germ-free and CRB-associated mice, which were housed in different isolators, were randomly grouped, and each group was fed with a γ-ray-sterilized CMF chow (Oriental Yeast) as a HFD, or AIN93G-fomula diet (Oriental Yeast) as a LFD for 4 weeks. To generate B.-thetaiotaomicron-associated mice, male or female germ-free mice of 8–16 weeks of age were colonized with a single gavage of 108 colony-forming units of B. thetaiotaomicron, JCM 5827T (Japan Collection of Microorganisms). Ten days after the inoculation, B.-thetaiotaomicron-associated mice were randomly grouped and each group was fed with the diet containing 15% (w/w) HAMS and HAMSB as a substitute for corn starch for 4 weeks. C57BL/6 mice and Myd88−/−Ticam1−/− mice were purchased from CLEA Japan and Oriental Bioservice, respectively. Mice were fed with AIN93G as a control diet for a week, and subsequently a diet containing 15% (w/w) HAMS, HAMSA, HAMSP or HAMSB for 4 weeks. Foxp3-hCD2-hCD52 knock-in (Foxp3hCD2 ) mice were described previously29. OT-II transgenic mice were from the Jackson Laboratory. All animal experiments were performed using protocols approved by Animal Studies Committees of RIKEN Yokohama Institute and Yokohama City University. Sample size was determined based on published studies using similar assays as well as the previous experience of the senior authors. No blinding was done for animal studies.
Adoptive transfer of OT-II T cells
OT-II (Ly5.2) transgenic CD4+ T cells were enriched from the spleen and lymph nodes using the IMag Cell Separation System. CD3+CD4+CD25−Vα2+ cells were sorted using FACSAriaII and intravenously transferred into C57BL/6 (Ly5.1) mice. Recipient mice were administered 1% ovalbumin in drinking water for 9 days.
Online Methods
Animals
Germ-free IQI mice were housed in germ-free isolators in the animal facility of RIKEN. Eight-to-ten-week-old male or female germ-free mice were orally inoculated with 3% (v/v) chloroform-treated murine faecal suspension (CRB)6. Germ-free and CRB-associated mice, which were housed in different isolators, were randomly grouped, and each group was fed with a γ-ray-sterilized CMF chow (Oriental Yeast) as a HFD, or AIN93G-fomula diet (Oriental Yeast) as a LFD for 4 weeks. To generate B.-thetaiotaomicron-associated mice, male or female germ-free mice of 8–16 weeks of age were colonized with a single gavage of 108 colony-forming units of B. thetaiotaomicron, JCM 5827T (Japan Collection of Microorganisms). Ten days after the inoculation, B.-thetaiotaomicron-associated mice were randomly grouped and each group was fed with the diet containing 15% (w/w) HAMS and HAMSB as a substitute for corn starch for 4 weeks. C57BL/6 mice and Myd88−/−Ticam1−/− mice were purchased from CLEA Japan and Oriental Bioservice, respectively. Mice were fed with AIN93G as a control diet for a week, and subsequently a diet containing 15% (w/w) HAMS, HAMSA, HAMSP or HAMSB for 4 weeks. Foxp3-hCD2-hCD52 knock-in (Foxp3hCD2 ) mice were described previously29. OT-II transgenic mice were from the Jackson Laboratory. All animal experiments were performed using protocols approved by Animal Studies Committees of RIKEN Yokohama Institute and Yokohama City University. Sample size was determined based on published studies using similar assays as well as the previous experience of the senior authors. No blinding was done for animal studies.
Adoptive transfer of OT-II T cells
OT-II (Ly5.2) transgenic CD4+ T cells were enriched from the spleen and lymph nodes using the IMag Cell Separation System. CD3+CD4+CD25−Vα2+ cells were sorted using FACSAriaII and intravenously transferred into C57BL/6 (Ly5.1) mice. Recipient mice were administered 1% ovalbumin in drinking water for 9 days.
Flow cytometry
The following monoclonal antibodies were conjugated with biotin, FITC, AlexaFluor 488, phycoerythrin (PE), PerCP-Cy5.5, PE-Cy7, allophycocyanin (APC), AlexaFluor 647, Alexa700, APC-H7, eFluor450, Brilliant Violet 421 or V500 (from BD Bioscience, eBioscience or Biolegend): anti-human CD2 (RPA-2.10), anti-mouse CD3ε (145-2C11), CD4 (GK1.5), CD25 (PC61), CD44 (IM7), CD45R/B220 (RA3-6B2), CD45RB (16A), CD62L (MEL-14), CD103 (2E7), Gr1 (RB6-8C5), folate receptor 4, FR4 (eBio12A5), Foxp3 (FJK-16 s), Helios (22F6), IL-10 (JES5-16E3) and TER119 (TER-119). Biotinylated anti-mouse/rat neuropilin-1 polyclonal antibodies were from R&D Systems.
For intracellular staining of Foxp3 and Helios, lymphocytes were pre-incubated with FcγR (CD16/CD32)-blocking monoclonal antibody (93; eBioscience) before staining for surface antigens. The cells were then fixed, permeabilized and stained with relevant monoclonal antibodies using the Foxp3 staining set (eBioscience) according to the manufacturer’s instructions.
For intracellular IL-10 staining, lamina propria lymphocytes were cultured for 6 h in complete medium (RPMI1640 containing 10% FCS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, 55 μM mercaptoethanol and 20 mM HEPES, pH 7.2) supplemented with 50 ng ml−1 phorbol myristate acetate, 500 ng ml−1 ionomycin, and Golgi Plug (BD Bioscience) in 12-well plates. The lymphocytes were then stained with monoclonal antibodies against CD3ε, CD4, CD25 and FR4, followed by intracellular staining for IL-10 using a Cytofix/Cytoperm kit (BD Bioscience). Our preliminary experiments demonstrated that the colonic CD3ε+CD4+CD25+FR4+ population exclusively consists of Foxp3+ cells, consistent with a previous report30. The stained samples were analysed using FACSCanto II and FACSAria II flow cytometers with DIVA software (BD Biosciences) and FlowJo software version 9.3.2 (Tomy Digital Biology).
Preparation of lymphocytes
Colonic lamina propria lymphocytes were prepared as described previously31. In brief, colonic tissues were treated with HBSS (Wako Pure Chemical industries) containing 1 mM dithiothreitol and 20 mM EDTA at 37 °C for 20 min to remove epithelial cells. The tissues were then minced and dissociated with collagenase solution containing 0.5 mg ml−1 collagenase (Wako Pure Chemical Industries), 0.5mg ml−1 DNase I (Roche Diagnostics), 2% FCS, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin and 12.5 mM HEPES, pH 7.2, in RPMI 1640 medium (Sigma-Aldrich) at 37 °C for 30 min to obtain single-cell suspensions. After filtering, the single-cell suspensions were washed with 2% FCS in RPMI 1640, and subjected to Percoll gradient separation. The spleen and mesenteric lymph nodes were mechanically disrupted into single-cell suspensions.
In vivo EdU-incorporation assay
To detect proliferating cells in vivo, HAMS- or HAMSB-fed mice received intraperitoneal injection of 3 mg EdU in 200 μl PBS, followed by administration of drinking water containing 0.8 mg ml−1 EdU for 2 days before the analysis. EdU-incorporated cells in cLP were visualized using Click-it EdU Flow cytometry kit (Invitrogen), following the manufacturer’s instructions.
Microbiological analysis
Bacterial genomic DNA was isolated as described previously with some modifications32. In brief, faecal samples were lyophilized by using VD-800R lyophilizer (TAITEC) for 24 h. Freeze-dried faeces were disrupted with 3.0 mm Zirconia Beads (Biomedical Science) by vigorous shaking (1,500 r.p.m. for 10 min) using Shake Master (Biomedical Science). Faecal samples (10 mg) were suspended with DNA extraction buffer containing 400 μl 10% (w/v) SDS/TE (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) solution, 400 μl phenol/chloroform/isoamyl alcohol (25:24:1), and 200 μl 3 M sodium acetate. Faeces in mixture buffer were further disrupted with 0.1 mm zirconia/silica beads (BioSpec Products) by vigorous shaking (1,500 r.p.m. for 10 min) using Shake Master (Biomedical Science). After centrifugation at 15,000g for 30 min at room temperature, bacterial genomic DNA was purified from the faecal extracts by a phenol/chloroform/isoamyl alcohol method. Adequate amounts of bacterial genomic DNA samples were amplified by the SYBR premix Ex Taq (TAKARA BIO) with the universal primers or Clostridiales-specific primers for the genes encoding the bacterial 16S rRNA sequence33. The results were calculated as the quantity relative to the copy number detected in the faeces of conventional mice.
454-barcoded pyrosequencing of 16S rRNA genes
The V1–V2 region of the 16S rRNA gene was amplified as described elsewhere34. Mixed samples were prepared by pooling approximately equal amounts of PCR amplicons from each sample and subjected to 454 GS JUNIOR (Roche Applied Science) sequencing according to the manufacturer’s instructions. 16S rRNA reads were analysed using QIIME(v1.6): fasta, quality files and a mapping file indicating the bar-code sequence corresponding to each sample were used as input. The QIIME pipeline takes this input information and split reads by samples according to the bar code, and classifies all 3,000 filter-passed reads of the 16S V1–V2 sequences obtained from each sample into operational taxonomic units on the basis of sequence similarity. It also performs taxonomical classification using the RDP-classifier (v2.5)35. The phylogenetic tree was constructed using ClustalW 2.0 (ref. 36) with 100 bootstrap iterations. The tree was visualized with iTOL37.
NMR-based metabolomics
Faecal and caecal metabolites were extracted by gentle shaking with 100 mM potassium phosphate buffer containing 90% deuterium oxide and 1 mM sodium 2,2-dimethyl-2- silapentane-5-sulphonate as the chemical shift reference (δ = 0.0 p.p.m.), and then analysed by 1H-NMR and 1H, 13C-NMR (refs 38, 39). PCA and orthogonal partial least squares discriminate analysis on the caecal metabolome data were run with the SIMCA-P+ software (ver 12.0, Umetrics)40,41. All NMR experiments were conducted using Bruker DRX-700 spectrometer equipped with a cryogenically cooled probe. The NMR spectra were processed essentially as described42,43,44. In brief, 1H-NMR data were reduced by subdividing the spectra into sequential 0.04 p.p.m. designated regions between 1H chemical shifts of 0.0 to 9.5 p.p.m. After exclusion of water resonance, each region was integrated and normalized to the total of all resonance integral regions. Metabolite annotations were performed using our standard database45,46.
Mass spectrometry
Organic acid concentrations of caecal contents and colonic tissues were determined by gas chromatography–mass spectrometer (GC–MS)47. In brief, 10 mg caecal contents or colonic tissues were disrupted using 3 mm zirconia/silica beads (BioSpec Products) and homogenized with extraction solution containing 100 μl of internal standard (100 μM crotonic acid), 50 μl HCl and 200 µl ether. After vigorous shaking using Shakemaster neo (Bio Medical Science) at 1,500 r.p.m. for 10 min, homogenates were centrifuged at 1,000g for 10 min and then the top ether layer was collected and transferred into new glass vials. Aliquots (80 μl) of the ether extracts were mixed with 16 μl N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA). The vials were sealed tightly, heated at 80 °C for 20 min in a water bath, and then left at room temperature for 48 h for derivatization. The derivatized samples were run through a 6890N Network GC System (Agilent Technologies) equipped with HP-5MS column (0.25 mm × 30 m × 0.25 μm) and 5973 Network Mass Selective Detector (Agilent Technologies). Pure helium (99.9999%) was used a carrier gas and delivered at a flow rate of 1.2 ml min−1. The head pressure was set at 97 kPa with split 20:1. The inlet and transfer line temperatures were 250 and 260 °C, respectively. The following temperature program was used: 60 °C (3 min), 60–120 °C (5 °C per min), 120–300 °C (20 °C per min). One microlitre of each sample was injected with a run time of 30 min. Organic acid concentrations were quantified by comparing their peak areas with the standards.
HPLC analysis of faecal organic acids
Faecal and caecal organic acids were measured by the internal standard method using an HPLC equipped with Shim-pack SCR-102H column (Shimadzu) and an electroconductivity detector (CDD-6A, Shimadzu)48.
In vitro cultures
CD4+ cells were enriched from the spleen and lymph nodes of C57BL/6 mice by a negative selection method with the IMag Cell Separation System (BD Bioscience) using a mixture of biotinylated monoclonal antibodies against B220, CD8α, CD11c, Gr-1 and Ter-119 and Streptavidin Particle Plus-DM (all from BD Bioscience). The enriched CD4+ fraction was subjected to cell sorting with FACSAriaII to isolate CD3+CD4+CD25−CD44loCD62Lhinaive T cells. For Treg cell polarization, naive CD4+ T cells (5 × 105 cells ml−1) were stimulated with immobilized anti-CD3 monoclonal antibody (10 μg ml−1) and soluble anti-CD28 monoclonal antibody (1 μg ml−1) supplemented with 0.2 ng ml−1 TGF-β1 and 10 ng ml−1 IL-2 (both from R&D Systems) in the presence or absence of 0.1 mM sodium acetate, sodium propionate or sodium butyrate (Sigma-Aldrich) for 1–3 days (Fig. 3). In the experiments described in Fig. 2a, b and Supplementary Fig. 7, T cells (5 × 105 cells ml−1) were cultured for 3 days and then expanded in complete medium supplemented with TGF-β and IL-2 for 2 days followed by analyses. T cells were polarized to TH1, TH2 or TH17 subtypes in vitro by the addition of the following: 10 ng ml−1 IL-12 (R&D Systems) and 10 μg ml−1 anti-IL-4 monoclonal antibody (clone 11B11) for TH1 cell polarization; 10 ng ml−1 IL-4 (R&D Systems) and 10 μg ml−1 anti-IL-12 monoclonal antibody (clone: R4-6A2) for TH2 cell polarization; and 40 ng ml−1 IL-6 (R&D Systems) and 0.2 ng ml−1 TGF-β1, along with 10 μg ml−1 each of anti-IFN-γ and anti-IL-4 monoclonal antibodies for TH17 cell polarization. IL-2 was added to the medium 48 h after initial stimulation, and T cell were cultured for 2 days followed by analyses.
ChIP
ChIP assays were performed using the MAGnify ChIP system (Invitrogen) according to the manufacturer’s protocol with a few modifications. In brief, 500,000 cells were fixed with 1% formaldehyde (room temperature, 10 min) and the reaction was stopped by the addition of glycine to a final concentration of 125 mM. Crude nuclei were isolated in SDS lysis buffer to obtain the chromatin solution. The chromatin solution was sonicated using a focused-ultrasonicator (Covaris) to reduce the chromatin DNA length to approximately 100–300 base pairs. The acoustic parameters were optimized (Duty cycle: 10%, Intensity: 5, Cycle/Burst: 200, 15 cycles) with monitoring the DNA fragment using Agilent 2100 Bioanalyzer. The sheared chromatin was immunoprecipitated (overnight, 4 °C) with magnetic protein A/G beads with immobilized 3 μg of anti-acetyl histone H3 antibody (Millipore) or rabbit IgG on a rotating wheel. After extensive washing, immune complexes were eluted (55 °C, 30 min) and reverse crosslinking was carried out (65°C, 1 h) in the presence of proteinase K. qPCR analysis was performed following magnetic bead-based DNA purification. For ChIP-seq library construction, immunoprecipitated DNA fragments were ligated with adaptors and amplified by PCR. Amplified libraries were subjected to cluster generation and sequencing analysis with the HiSeq 1000 (Illumina). Sequenced reads were mapped to the mouse genome (v. mm9) with BOWTIE. Peaks for each population were called with MACS with P value threshold of less than 1 × 10−5. The numbers of reads in a section were that of total mapped reads and the length of each section. Because butyrate treatment globally enhances histone acetylation, total mapped reads may be increased in butyrate-treated cells compared to control cells. Therefore, the normalized values within each sample were further subjected to cross-sample adjustment on the basis of read counts of the four genomic regions around transcription start sites of the housekeeping gene Rpl13a, whose histone acetylation status remained unchanged between butyrate-treated and untreated cells as confirmed by ChIP-qPCR analysis (data not shown). The normalized reads were calculated within fixed base length for upstream and downstream regions, and relative length for genic regions.
Gene expression profiling
Total RNA was extracted by Trizol reagent (Life Technologies) following a standard protocol, and subjected to microarray analysis using the GeneChip Mouse Gene 1.0 ST Array (Affymetrix). Microarray signals were processed using a standard robust multiarray averaging (RMA) algorithm. Observed signals were normalized using quantile normalization method and genes that had no significant signals were ignored to reduce noise. Butyrate-sensitive transcription factors were selected using annotation provided by Gene Ontology, as they have annotation ‘nucleus (GO:0005634)’ and ‘regulation of transcription, DNA-dependent (GO:0006355)’. Their acetylation was represented by the normalized peak height of ChIP-seq reads from 4 kb upstream to 4 kb downstream of the transcription start site. The distribution of log ratio between control and butyrate treatment could be approximated with two Gaussian distributions and fitted curves were determined using expectation–maximization algorithm. The threshold value was log2 0.5.
Induction of colitis by adoptive transfer of CD4+CD45RBhigh T cells
Colitis was induced in Rag1−/− mice by adoptive transfer of CD4+CD45RBhigh T cells as described previously21. In brief, CD4+ T cells were enriched from splenocytes from C57BL/6 mice by IMag Cell Separation System. Enriched CD4+ T cells were labelled with FITC-conjugated anti-mouse CD3ε (145-2C11), PE-conjugated anti-mouse CD45RB (16A) (all from BD Biosciences), and CD3ε+CD4+CD45RBhigh cells were isolated by cell sorting using FACSAria II flow cytometer (BD Biosciences). The Rag1−/− recipients were given 2 × 105 CD4+CD45RBhigh T cells via the tail vein, and were euthanized at 6 weeks after transfer. For the Treg cell depletion experiment, CD4+ CD45RBhigh T cells from Foxp3hCD2 reporter mice were adoptively transferred into Rag1−/− mice. The recipient mice were received intravenous injection of 1 mg anti-human CD2 monoclonal antibody (35.1) at 4 and 5 weeks after the transfer.
Histology
Prefixed colonic tissue sections were deparaffinized, rehydrated and stained with haematoxylin and eosin or with Alcian blue-nuclear fast red. The specimens were examined histologically for scoring the degree of colitis based on the following criteria: inflammatory infiltrates, mucosal hyperplasia and loss of goblet cells.
Immunofluorescent staining
Immunofluorescent staining of cross-sections of colonic tissues was performed as described previously49.
Immunoblotting
Immunoblotting analysis was performed as described previously50. Antibodies against histone H3 (ab1791, Abcam), acetylated histone H3 (06-599, Merck), Stat5 (clone: 3H7, Cell Signaling Technology), Smad3 (clone: C67H9, Cell Signaling Technology), NFAT1 (4389, Cell Signaling Technology), c-Rel (AF2699, R&D systems) and GAPDH (clone: 6C5, MAB374, Merck) were used in this study.
Statistical analyses
Differences between two or more groups were analysed respectively by the Student’s t-test or ANOVA followed by Tukey’s post-hoc test. When variances were not homogeneous, the data were analysed by the non-parametrical Mann–Whitney U test or the Kruskal–Wallis test followed by the Scheffé test. The body weight changes of the mice fed with or without HAMSB were analysed using a two-way repeated measures ANOVA followed by Tukey’s test. All statements indicating significant differences show at least a 5% level of probability.
Accession codes
Accessions
DDBJ/GenBank/EMBL
Gene Expression Omnibus
Data deposits
The microarray and ChIP-seq analysis data have been deposited at the Gene Expression Omnibus (GEO) under accession number GSE49655. The microbiome analysis data have been deposited at the DDBJ database (http://getentry.ddbj.nig.ac.jp/) under accession number DRA001105.
Change history
12 February 2014
An Erratum to this paper has been published: https://doi.org/10.1038/nature13041
References
Chung, H. et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593 (2012)
Ivanov, I. I. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009)
Atarashi, K. et al. Induction of colonic regulatory T cells by indigenous clostridium species. Science 331, 337–341 (2011)
Geuking, M. B. et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 34, 794–806 (2011)
Round, J. L. & Mazmanian, S. K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl Acad. Sci. USA 107, 12204–12209 (2010)
Itoh, K. & Mitsuoka, T. Characterization of clostridia isolated from faeces of limited flora mice and their effect on caecal size when associated with germ-free mice. Lab. Anim. 19, 111–118 (1985)
Thornton, A. M. et al. Expression of Helios, an Ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 184, 3433–3441 (2010)
Yadav, M. et al. Neuropilin-1 distinguishes natural and inducible regulatory T cells among regulatory T cell subsets in vivo . J. Exp. Med. 209, 1713–1722 (2012)
Weiss, J. M. et al. Neuropilin 1 is expressed on thymus-derived natural regulatory T cells, but not mucosa-generated induced Foxp3+ T reg cells. J. Exp. Med. 209, 1723–1742 (2012)
Annison, G., Illman, R. J. & Topping, D. L. Acetylated, propionylated or butyrylated starches raise large bowel short-chain fatty acids preferentially when fed to rats. J. Nutr. 133, 3523–3528 (2003)
Rubtsov, Y. P. et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 28, 546–558 (2008)
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by Toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004)
Mazmanian, S. K., Round, J. L. & Kasper, D. L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 453, 620–625 (2008)
Candido, E. P., Reeves, R. & Davie, J. R. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell 14, 105–113 (1978)
Davie, J. R. Inhibition of histone deacetylase activity by butyrate. J. Nutr. 133, 2485S–2493S (2003)
de Zoeten, E. F., Wang, L., Sai, H., Dillmann, W. H. & Hancock, W. W. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 138, 583–594 (2010)
Tao, R. et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nature Med. 13, 1299–1307 (2007)
Josefowicz, S. Z., Lu, L.-F. & Rudensky, A. Y. Regulatory T cells: mechanisms of differentiation and function. Annu. Rev. Immunol. 30, 531–564 (2012)
Zheng, Y. et al. Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812 (2010)
Ruan, Q. et al. Development of Foxp3+ regulatory T cells is driven by the c-Rel enhanceosome. Immunity 31, 932–940 (2009)
Powrie, F., Leach, M. W. M., Mauze, S. S., Caddle, L. B. L. & Coffman, R. L. R. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int. Immunol. 5, 1461–1471 (1993)
Maslowski, K. M. et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286 (2009)
Brown, A. J. et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 278, 11312–11319 (2003)
Inan, M. S. et al. The luminal short-chain fatty acid butyrate modulates NF-κB activity in a human colonic epithelial cell line. Gastroenterology 118, 724–734 (2000)
Thibault, R. et al. Down-regulation of the monocarboxylate transporter 1 is involved in butyrate deficiency during intestinal inflammation. Gastroenterology 133, 1916–1927 (2007)
Frank, D. N. et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl Acad. Sci. USA 104, 13780–13785 (2007)
Scheppach, W. et al. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology 103, 51–56 (1992)
Harig, J. M., Soergel, K. H., Komorowski, R. A. & Wood, C. M. Treatment of diversion colitis with short-chain-fatty acid irrigation. N. Engl. J. Med. 320, 23–28 (1989)
Miyao, T. et al. Plasticity of Foxp3+ T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity 36, 262–275 (2012)
Yamaguchi, T. et al. Control of immune responses by antigen-specific regulatory T cells expressing the folate receptor. Immunity 27, 145–159 (2007)
Weigmann, B. et al. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nature Protocols 2, 2307–2311 (2007)
Date, Y. et al. New monitoring approach for metabolic dynamics in microbial ecosystems using stable-isotope-labeling technologies. J. Biosci. Bioeng. 110, 87–93 (2010)
Bouskra, D. et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510 (2008)
Kim, S. W. et al. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 20, 241–253 (2013)
Reyes, A. et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 466, 334–338 (2010)
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007)
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128 (2006)
Fukuda, S. et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 469, 543–547 (2011)
Fukuda, S. et al. Evaluation and characterization of bacterial metabolic dynamics with a novel profiling technique, real-time metabolotyping. PLoS ONE 4, e4893 (2009)
Kruger, N. J., Troncoso-Ponce, M. A. & Ratcliffe, R. G. 1H NMR metabolite fingerprinting and metabolomic analysis of perchloric acid extracts from plant tissues. Nature Protocols 3, 1001–1012 (2008)
Wiklund, S. et al. Visualization of GC/TOF-MS-based metabolomics data for identification of biochemically interesting compounds using OPLS class models. Anal. Chem. 80, 115–122 (2008)
Kikuchi, J., Shinozaki, K. & Hirayama, T. Stable isotope labeling of Arabidopsis thaliana for an NMR-based metabolomics approach. Plant Cell Physiol. 45, 1099–1104 (2004)
Tian, C. et al. Top-down phenomics of Arabidopsis thaliana: metabolic profiling by one- and two-dimensional nuclear magnetic resonance spectroscopy and transcriptome analysis of albino mutants. J. Biol. Chem. 282, 18532–18541 (2007)
Sekiyama, Y., Chikayama, E. & Kikuchi, J. Profiling polar and semipolar plant metabolites throughout extraction processes using a combined solution-state and high-resolution magic angle spinning NMR approach. Anal. Chem. 82, 1643–1652 (2010)
Akiyama, K. et al. PRIMe: a Web site that assembles tools for metabolomics and transcriptomics. In Silico Biol. 8, 339–345 (2008)
Chikayama, E. et al. Statistical indices for simultaneous large-scale metabolite detections for a single NMR spectrum. Anal. Chem. 82, 1653–1658 (2010)
Sannasiddappa, T. H., Costabile, A., Gibson, G. R. & Clarke, S. R. The influence of Staphylococcus aureus on gut microbial ecology in an in vitro continuous culture human colonic model system. PLoS ONE 6, e23227 (2011)
Morita, T. et al. Resistant proteins alter cecal short-chain fatty acid profiles in rats fed high amylose cornstarch. J. Nutr. 128, 1156–1164 (1998)
Obata, Y. et al. Epithelial cell-intrinsic Notch signaling plays an essential role in the maintenance of gut immune homeostasis. J. Immunol. 188, 2427–2436 (2012)
Furusawa, Y. et al. DNA double-strand breaks induced by cavitational mechanical effects of ultrasound in cancer cell lines. PLoS ONE 7, e29012 (2012)
Acknowledgements
We would like to thank P. Carninci, Y. Shinkai and M. Yoshida for discussion; Y. Chiba and S. Yamada for technical support; H. Sugahara for technical advice; and P. D. Burrows for critical reading and editing of the manuscript. This work was supported in part by grants from Japanese Ministry of Education, Culture, Sports, Science and Technology (24117524 to S.F.; 21022049 to K.Ha.; 20113003 to H.O.), The Japan Society for the Promotion of Science (24890293 to Y.F.; 252667 to Y.O.; 24380072 and 24658129 to S.F.; 22689017 to K.Ha.; 21390155 to H.O.), The Japan Science and Technology Agency (K.Ha., K.A. and K.Ho.), RIKEN President’s Special Research Grant (H.O.), RIKEN RCAI Young Chief Investigator program (K.Ha.), the Institute for Fermentation, Osaka (S.F.), the Mishima Kaiun Memorial Foundation (S.F.), The Takeda Science Foundation (S.F. and H.O.), The Mitsubishi Foundation (H.O.), and The Uehara Memorial Foundation (S.F. and K.Ha.).
Author information
Authors and Affiliations
Contributions
S.F., K.Ha., D.L.T., T.M., K.Ho. and H.O. conceived the study; K.Ha. and S.F. designed the experiments and wrote the manuscript with Y.Fur., Y.O. and H.O.; Y. Fur. and Y.O conducted a large part of experiments together with S.F., G.N., D.T., C.U., K.K., T.K., M.Ta., E.M. and K.Ha; S.F, S.O. and K.Ha. prepared germ-free, CRB-associated and gnotobiotic mice. K.A. and K.Ho. were involved in data discussion. S.F., Y.N., C.U. and J.K. performed metabolome analysis. S.F., T.K., S.M. and M.To. performed microbiome analysis. T.A.E. performed bioinformatic analyses. S.Hi. and T.M. performed HPLC analysis. S.F. and N.N.F. performed GC–MS analysis. Y.Fuj. performed histological analysis. T.L., J.M.C., D.L.T. and S.Ho. provided essential materials and contributed to the design of experiments. Y.Fur. and H.K. contributed to the ChIP assay. H.O. directed the study and took primary responsibility for editing the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
This file contains Supplementary Figures 1-22 and Supplementary Table 1. (PDF 2523 kb)
Rights and permissions
About this article
Cite this article
Furusawa, Y., Obata, Y., Fukuda, S. et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450 (2013). https://doi.org/10.1038/nature12721
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature12721
This article is cited by
-
Gut microbial metabolites SCFAs and chronic kidney disease
Journal of Translational Medicine (2024)
-
Causal relationship between gut microbiota and gastrointestinal diseases: a mendelian randomization study
Journal of Translational Medicine (2024)
-
Metabolic network of the gut microbiota in inflammatory bowel disease
Inflammation and Regeneration (2024)
-
Targeted metabolomics reveals plasma short-chain fatty acids are associated with metabolic dysfunction-associated steatotic liver disease
BMC Gastroenterology (2024)
-
Association of gut microbiota with the pathogenesis of SARS-CoV-2 Infection in people living with HIV
BMC Microbiology (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
